(If anything starts to look too unfamiliar, review Thermochemistry, Chapter 6)
Thermodynamics is a “dead” science; it's a complete science. Because of this, we can test every new hypothesis against it, because it is true. It is the only true test.
Thermodynamics works even if matter is continuous (if matter can be halved and halved and halved forever).
Terms:
System: Whatever we're studying, things that produce the energy
Surroundings: Everything else in the universe
There are the 1st, 2nd, 3rd, and 0th Laws.
0th Law of Thermodynamics: Same as the Reflexive Property of Equality for mathematics:
a = a; 3 = 3.
Imagine three closed systems in thermal equilibrium (A, B, and C). If the temperature of A equals the temperature of B, and if the temperature of B equals the temperature of C, then the temperature of A equals the temperature of C.
In other words: Thermometers work!
First Law of Thermodynamics: Law of Conservation of Energy
Total energy is constant. Energy is neither created nor destroyed. “You never get more than you pay for.” Put a certain amount in, you'll never get more out.
Internal Energy: Denoted U, is the sum of the kinetic and potential energies of the particles making up the system. Kinetic energy = energy of motion of electrons, nuclei and molecules. Potential energy = bonds in the molecules and attractions between them.
U = internal energy = PE + KE
U is relative (there is no true zero of positional energy)
ΔU: the change is what's important
Change in Internal Energy equals heat plus work → ΔU = q + w
Thermo = Heat = q
Dynamics = Work = w
Heat: energy that moves into or out of the system because of a temperature difference between the system and its surroundings. In this case, due to random motion. q units are kJ (kilojoules). Heat flows in one direction: hot to cold
heat enters the system → +q
heat leaves the system → – q
Remember from Chapter 6:
q = s x m x Δt
(s = specific heat, m = mass, Δt = change in temperature)
qp = heat at constant pressure
qp = ΔH → heat of reaction at constant pressure is the same as change in enthalpy
Work: the energy exchange that results when a force F moves an object through a distance d.
w = F x d
– w ; work done on the surroundings by the system
+ w; work done on the system by the surroundings.
Example: Piston-cylinder system:
Zn + 2HCl → ZnCl2 + H2
The constant pressure of the atmosphere is replaced by the piston and weight. As hydrogen gas evolves and pushes up against the piston, work is done by the system on the surroundings. This is represented as – w. Or:
w = – F x Δh ← (distance is represented by the change in height the piston moves in the cylinder)
Pressure = Force / Surface Area
*Solve this expression for Force:
Force = Pressure x Surface Area = P x A
*Replace this expression for Force in the first equation:
w = – P x A x Δh
*Area times height is the same as volume. Replace A x Δh with ΔV for change in volume.
w = – PΔV → “PV work”
w = – P(vf – vi)
In other words: you can calculate the work done by a chemical reaction carried out in an open vessel by multiplying the atmospheric pressure by the change in volume of the chemical system.
Review:
ΔU = q + w
qp = heat at constant pressure
w = – PΔV
ΔU = qp – PΔV
qp = ΔH
ΔU = ΔH – PΔV
Example:
qp for a reaction is – 511 kJ (heat went out). Pressure = 2.05 x 105 Pa. Initial Volume = 5.05 m3. Final Volume = 6.15 m3.
A note on units:
Pa x m3 = J
ΔU = J
w = J
– P = Pa
101,325 Pa = 1 atm
ΔV = m3
What is the internal energy?
ΔU = qp – PΔV
ΔU = – 511 kJ – (2.05 x 105 Pa)(6.15 m3 – 5.05 m3)
ΔU = – 511 kJ – 225500J → convert J to kJ by dividing by 1000
ΔU = – 511 kJ – 226 kJ = – 737 kJ
Example:
For a reaction in a piston/cylinder system, the heat at constant pressure was – 815 kJ, and the volume contracted from 3.15 m3 to 2.50 m3. Pressure = 1.56 x 105 Pa. What is the ΔU for this process?
ΔU = – 815 kJ – (1.56 x 105 Pa)(2.50 m3 – 3.15 m3)
ΔU = – 815 kJ – (– 101400 J)
ΔU = – 815 kJ + 101 kJ = – 714 kJ
From ΔU = ΔH – PΔV, rearrange to get:
ΔH = ΔU + PΔV
*If there is no change in volume (or a negligible change) then
ΔH = ΔU (because PΔV would be zero)
For example, a bomb calorimeter does not allow a change in volume. At constant volume, ΔH = ΔU.
qwhatever means constant whatever:
qv = heat at constant volume = ΔU
qp = heat at constant pressure = ΔH
State Functions: Do not depend on the history of the system. All we care about is initial and final values.
ΔH = Hf – Hi → state function
Example: Route up a hill, the change in altitude you experience from the bottom to the top of the hill is the same no matter what route you take to get to the top of the hill.
ΔH, ΔU, ΔT, ΔV, ΔS, ΔG → all state functions
q and w are NOT state functions.
But qp, qv, and (q + w) are state functions
Example of the First Law of Thermodynamics:
Perpetual Motion DEVICES:
Are possible because devices don't do work.
Perpetual Motion MACHINES:
Are not possible because machines do work. The machine stops doing work because it eventually runs out of energy. Without an input of energy, because of the first law, you can't get more than you pay for.
Second Law of Thermodynamics:
“You get cheated every time.” Energy gets lost as random motion (frictional heat). The total entropy of a system and its surroundings always increases for a spontaneous process.
Spontaneous vs. Non-Spontaneous Processes:
Spontaneous Process: a physical or chemical change that occurs by itself, without any outside force to help it along.
Non-Spontaneous Process: the opposite of a spontaneous process. A process that does not naturally occur all by itself.
-A ball rolling down a hill = spontaneous.
-A ball rolling up a hill = non-spontaneous (not that it's impossible, it's just highly improbable). The ball could be rolled up the hill, but it would require work.
Most processes are “possible” , just so very unlikely that they'll just about never happen.
Example: You scatter a deck of cards randomly, face down on a table. The odds of you picking them up in perfect order is 1 in 52! (factorial), or 1 in 8.07 x 1067. It is not impossible, it is just highly unlikely.
Dependent on an idea called entropy.
Entropy: Denoted S, is randomness. A thermodynamic quantity representing the amount of energy in a system that is no longer available for doing mechanical work; "entropy increases as matter and energy in the universe degrade to an ultimate state of inert uniformity." Entropy represents “energy dispersal”, not specifically energy.
Because you want one possibility out of many, you're fighting randomness. (This is why we age.)
The entropy of the universe increases with every spontaneous process.
Entropy = S = in units J / K (Joules / Kelvin)
Suniverse = Ssurroundings + Ssystem
ΔS = Sf – Si
ΔS = entropy created + (q / T)
The term “entropy created” equals the movement of molecules, breaking of bonds, etc. It is very hard to quantify, so it is ignored, and we use:
ΔS = (q / T)
q = random motion in units J
T = temperature in degrees Kelvin
The entropy change associated with a flow of heat, q, at an absolute temperature, T.
At constant pressure,
ΔS is greater than qp / T (which equals ΔH / T)
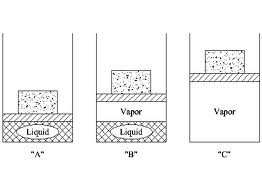
but at equilibrium (at phase change, going between solid and liquid, solid and gas, or liquid and gas)
ΔS = ΔH / T
but ΔS = ΔH / T exactly only when the phase change takes forever.
But as long as the phase change happens relatively slowly, this is a great approximation.
At equilibrium and at constant pressure:
ΔHfus = heat of fusion (liquid to solid; melting)
ΔHvap = heat of vaporization (liquid to gas)
ΔHcond = condensation (gas to liquid)
Example:
ΔHvap = +42.5 kJ / mol for SeF4.
If 1 mol of liquid SeF4 at 25oC has an entropy of 185 J / K, what is the entropy of 1 mol of SeF4 gas at equilibrium with the liquid?
*Everything with a temperature greater than 0 Kelvin has a positive entropy within itself (that is inherent).
ΔSgas = ΔSliquid + (ΔHvap / T)
As ΔS increases, the randomness increases.
ΔSliquid = 185 J / K
ΔHvap = 42.5 x 103 J
T = 298 K
ΔSgas = 185 J / K + (42.5 x 103 J / 298 K)
ΔSgas = 328 J / K
Example:
ΔHcond = – 58.5 kJ / mol for TeF4.
If 1 mol of gaseous TeF4 at 25oC has an entropy of 453 J / K, what is the entropy of 1 mol of TeF4 liquid at equilibrium with the gas?
ΔSliquid = ΔSgas – (ΔHcond / T)
ΔSliquid = 453 J / K – (58.5 x 103 J / 298 K)
ΔSliquid = 257 J / K
For processes at constant temperature and pressure:
ΔH – TΔS is less than zero when the process is spontaneous.
ΔH – TΔS is greater than zero when the process is non-spontaneous.
This process will be spontaneous in the opposite direction.
ΔH – TΔS is equal to zero when the reaction is at equilibrium.
Third Law of Thermodynamics:
A substance that is a perfect crystal (pure, no breaks or occlusions) at zero degrees Kelvin has 0 entropy. No randomness.
S0 K = 0 for any substance
ΔSo = So (standard conditions) = absolute entropy
This is the entropy value for the standard state, which is the pure substance at 1 atm pressure, or the substance in solution at a concentration of 1 M.
Qualitative Info From Equations on Entropy:
There is an increase in entropy when:
1. A reaction in which a molecule is broken into two smaller molecules.
2. A reaction in which there is an increase in moles of gas (moles of gas of product is greater than moles of gas of reactants).
3. A process in which a solid changes to liquid or gas, or a liquid changes to a gas.
Entropy rises gradually as the temperature increases but jumps sharply at each phase transition.

Example:
Is ΔSo positive or negative?
CaCO3 (s) → CaO (s) + CO2 (g)
ΔSo is positive, see Rule 1.
CS2 (g) → CS2 (l)
ΔSo is negative, see Rule 3.
2Hg (l) + O2 (g) → 2HgO (s)
ΔSo is negative, see Rule 1.
Note: gas formation wins out increasing in entropy.
Remember from chapter 6:
ΔHorxn = ΣmΔHof(products) – ΣnΔHof(reactants)
Example:
CaCO3 (s) → CaO (s) + CO2 (g)
ΔHof CaCO3 = 92.9 J / K • mol
ΔHof CaO = 38.21 J / K • mol
ΔHof CO2 = 213.7 J / K • mol
Plug values into the ΔHorxn equation:
[(1mol)(38.21 J / K • mol) + (1 mol)(213.7 J / K • mol)] – [(1 mol)(92.9 J / K • mol)] = 159 J / K
The standard change of entropy, ΔSo, can be found for a reaction in a similar way.
ΔSorxn = ΣmΔSo(products) – ΣnΔSo(reactants)
ΔG = free energy
If ΔG is less than zero, reaction is spontaneous.
If ΔG is greater than zero, reaction is non-spontaneous and doesn't happen.
Thus:
ΔG = wmax ← maximum work that any process can produce (but you never actually get there).
When ΔGo is a large negative number, more negative than – 10 kJ, then the reaction is spontaneous (mostly products).
When ΔGo is a large positive number, greater than 10 kJ, then the reaction is non-spontaneous (mostly reactants).
When ΔGo is between – 10 kJ and 10 kJ, there will be a mixture of reactants and products.
ΔG = – 0.0100 kJ / mol is spontaneous
ΔG = + 0.0100 kJ / mol is non-spontaneous
ΔGorxn = ΣmΔGof(products) – ΣnΔGof(reactants)
and
ΔG = ΔH – TΔS
Example:
CH4 (g) + 2O2 (g) ⇌ CO2 (g) + 2H2O (l)
**Note: ΔHof values in Table 6.2 on page 246, Sof values in Table 18.1 on page 743, and ΔGof values in Table 18.2 on page 747. Values are also together in Appendix C.**

Find ΔSo and ΔHorxn at 298 K.
Then use these values to find ΔG.
ΔHorxn = [(1 mol)(– 393.5 KJ / mol) + (2 mol)(– 285.8 KJ / mol)] – [(1 mol)(– 74.87 KJ / mol) + (2 mol)(0)] = – 890 KJ
ΔSo = [(1 mol)(213.7 J / K • mol) + (2 mol)(69.95 J / K • mol)] – [(1 mol)(186.1 J / K • mol) + (2 mols)(205.0 J / K • mol) = – 242.5 J / K
There are two methods to solve for ΔG:
ΔG = ΔH – TΔS
ΔG = (– 890 KJ) – (298 K)(– 242 J / K)
ΔG = (– 890 KJ) – (72.27 KJ) = – 817.7KJ
or plug values into the previously stated equation for ΔG:
ΔGorxn = ΣmΔGof(products) – ΣnΔGof(reactants)
When you solve for ΔG using these two methods, your answer will not be exactly the same, but very close.
Relationship Between Thermodynamics and Kinetics:
ΔG ↔ K (thermodynamic equilibrium constant)
ΔGorxn = – RTlnK
Where:
R = 8.31 J / K • mol (Ideal Gas Constant in J / K • mol)
T = Temperature in degrees Kelvin
K = Kc or Ka or Kb or Ksp or Kf or Kd, etc.
Example:
AgCl (s) ⇌ Ag+ (aq) + Cl- (aq)

Find K.
ΔGorxn = [(1 mol)(77.1 kJ) + (1 mol)(– 133.3 kJ)] – [(1 mol)(–109.8 kJ)] = 55.6 kJ = 55.6 x 103 J
55.6 x 103 J = – (8.31 J / K • mol)(298 K)(ln K)
(55.6 x 103) / [(– 8.31)(298)] = ln K
– 22.45 = ln K
e– 22.45 = K
K = 1.78 x 10–10
Example:
Ka = 1.7 x 10–5
Find ΔG.
ΔG = (– 8.31 J / K • mol)(298 K)(ln 1.7 x 10–5) = 27196.84 J = +27.2 kJ / mol
Consider the equation ΔG = ΔH – TΔS
How is ΔG affected by temperature?

ΔG = 0 is the point where reaction goes from spontaneous to non-spontaneous.
Very impressive and informative blog.Enthalpy is studied under Thermodynamics and its one of the major branch of science in which heat and change of heat is studied.
ReplyDelete
ReplyDeleteBest ias coaching in bangalore
.www.globalias.in
There is a typo in the last image. As you said yourself in "ΔG = 0 is the point where reaction goes from spontaneous to non-spontaneous," then the -ΔΗ and -ΔS row should say "Spontaneous at low temp. As temp increases, ΔG goes from negative to positive, so it becomes *non-spontaneous at high temps." (Correction in asterisk, *non-)
ReplyDelete